Polymer in water#
Solvating a small molecule in water before stretching it

The goal of this tutorial is to use GROMACS and create a small hydrophilic polymer (PEG - PolyEthylene Glycol) in a reservoir of water. An all-atom description is used, therefore all species considered here are made of charged atoms connected by bond constraints.
Once the system is created, a constant stretching force will be applied to both ends of the polymer, and its length will be measured with time.
This tutorial was inspired by a very nice publication by Liese and coworkers, in which they compare MD simulations of a PEG molecule in water with force spectroscopy experiments.
Looking for help with your project?
See the Contact page.
PEG molecule in vacuum#
Download the peg.gro file for the PEG molecule by clicking here.
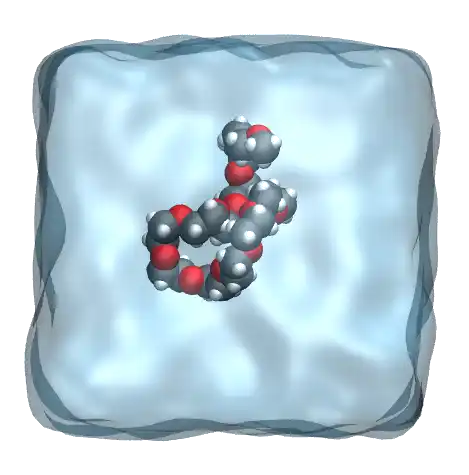
Opening peg.gro using VMD, one can see that it consists of a rather long polymer chain main of carbon atoms (in gray), oxygen atoms (in red), and hydrogen atoms (in white):

This PEG molecule contains 24 carbon atoms, 12 oxygen atoms, and 50 hydrogen atoms.#
Create a folder named free-peg-in-vacuum/, and copy peg.gro in it. Next to peg.gro create an empty file named topol.top, and copy the following lines in it:
[ defaults ]
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ
1 1 no 1.0 1.0
; Include forcefield parameters
#include "ff/charmm35r.itp"
#include "ff/peg.itp"
[ system ]
; Name
PEG
[ molecules ]
; Compound #mols
PEG 1
Next to conf.gro and topol.top, create a folder named ff/, and copy the following files into it: charmm35r.itp and peg.itp
These 2 files contain the parameters for the PEG molecules, as well as extra parameters for the water molecules that will be added later.
Create an inputs/ folder next to ff/, and create a new empty file called nvt.mdp. Copy the following lines into it:
integrator = md
dt = 0.002
nsteps = 500000
nstenergy = 1000
nstlog = 1000
nstxout = 1000
constraints = hbonds
coulombtype = pme
rcoulomb = 1.0
rlist = 1.0
vdwtype = Cut-off
rvdw = 1.0
tcoupl = v-rescale
tau_t = 0.1
ref_t = 300
tc_grps = PEG
gen_vel = yes
gen-temp = 300
gen-seed = 65823
comm-mode = angular
Run the simulation using GROMACS by typing in a terminal:
gmx grompp -f inputs/nvt.mdp -c peg.gro -p topol.top -o nvt -maxwarn 1
gmx mdrun -v -deffnm nvt
After the simulation is over, open the trajectory file with VMD by typing:
vmd peg.gro nvt.trr
You should see the PEG molecule moving due to thermal agitation.
Support me for 1 euro per month
Become a Patreon and support the creation of content for GROMACS
Angle distribution#
Let us use the power of GROMACS to extract the angular distribution between triplets of atoms during the run. First, let us create an index file from the peg.gro file:
gmx mk_angndx -s nvt.tpr -n index.ndx -hyd no
The first group created contains all the carbon and oxygen atoms (a total of 36 atoms), as can be seen from the index.ndx file:
[ Theta=109.7_795.49 ]
2 5 7 10 12 14 17 19 21 24 26 28
31 33 35 38 40 42 45 47 49 52 54 56
59 61 63 66 68 70 73 75 77 80 82 84
Here each number corresponds to the atom index, as can be seen from the peg.gro file. For instance, atom of id 2 is a carbon atom, and five in an oxygen:
PEG in water
86
1PEG H 1 2.032 1.593 1.545 0.6568 2.5734 1.2192
1PEG C 2 1.929 1.614 1.508 0.1558 -0.2184 0.8547
1PEG H1 3 1.902 1.721 1.523 -3.6848 -0.3932 -3.0658
1PEG H2 4 1.921 1.588 1.400 -1.5891 1.4960 0.5057
1PEG O 5 1.831 1.544 1.576 0.0564 -0.5300 -0.6094
1PEG H3 6 1.676 1.665 1.494 -2.6585 -0.5997 0.3128
1PEG C1 7 1.699 1.559 1.519 0.6996 0.0066 0.2900
1PEG H4 8 1.699 1.500 1.425 4.2893 1.6837 -0.9462
Using the index file, one can extract the angle distribution between all the species in the group Theta=109.7_795.49, by typing:
gmx angle -n index.ndx -f nvt.trr -od angdist.xvg -binwidth 0.25
Select the first group by typing 0. A file named angdist.xvg was created, it looks like it:
Angle distribution for the Carbon and oxygen atoms of the PEG molecule in vacuum.#
Pull on the PEG#
Let us apply a force and pull on the PEG polymer. Duplicate the free-peg-in-vacuum/ folder, and call the copy pulled-peg-in-vacuum/.
First, change the box size to make room for the pulling by replacing the last line of peg.gro from
3.00000 3.00000 3.00000
to
3.00000 3.00000 8.00000
Then, for convenience, let us center the PEG molecule in the box by typing:
gmx trjconv -f peg.gro -o peg-centered.gro -s nvt.tpr -center -pbc mol
And choose System for both centering and output. Then, let us specify to GROMACS which atoms are going to be pulled. This can be done by adding 2 additional groups named End1 and End2 to the index file index.ndx:
Create an index file by typing:
gmx make_ndx -f peg-centered.gro -o index.ndx
And create the 2 additional groups by typing:
a 82
a 5
name 3 End1
name 4 End2
Then press q to save and quit. The index file index.ndx contains 2 additional group, with one oxygen atom each:
(...)
[ PEG ]
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
16 17 18 19 20 21 22 23 24 25 26 27 28 29 30
31 32 33 34 35 36 37 38 39 40 41 42 43 44 45
46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
61 62 63 64 65 66 67 68 69 70 71 72 73 74 75
76 77 78 79 80 81 82 83 84 85 86
[ End1 ]
82
[ End2 ]
5
Then, duplicate the nvt.mdp file, call the duplicate pull.mdp.
Remove the comm-mode = angular line. Then, add the following lines
to pull.mdp:
pull = yes
pull-coord1-type = constant-force
pull-ncoords = 1
pull-ngroups = 2
pull-group1-name = End1
pull-group2-name = End2
pull-coord1-groups = 1 2
pull-coord1-geometry = direction-periodic
pull-coord1-dim = N N Y
pull-coord1-vec = 0 0 1
pull-coord1-k = 50
pull-coord1-start = yes
pull-print-com = yes
These lines are ensuring that a force is applied along the z direction to both groups End1 and End2. Turn off the velocity generator as well:
gen_vel = no
Re-equilibrate the system using the previous npt.mdp script, and then run the pull.mdp file:
gmx grompp -f inputs/nvt.mdp -c peg-centered.gro -p topol.top -o nvt -maxwarn 1
gmx mdrun -v -deffnm nvt
gmx grompp -f inputs/pull.mdp -c nvt.gro -p topol.top -o pull -n index.ndx
gmx mdrun -v -deffnm pull -px position.xvg -pf force.xvg
Here, the -n index.ndx command is used to refer to the previously created index file, so that GROMACS finds the End1 and End2 groups. The -px position.xvg and -pf force.xvg are used to print positions and forces of the 2 end groups in files.
Looking at the evolution of the position with time, one can see that the polymer stretches very quickly:
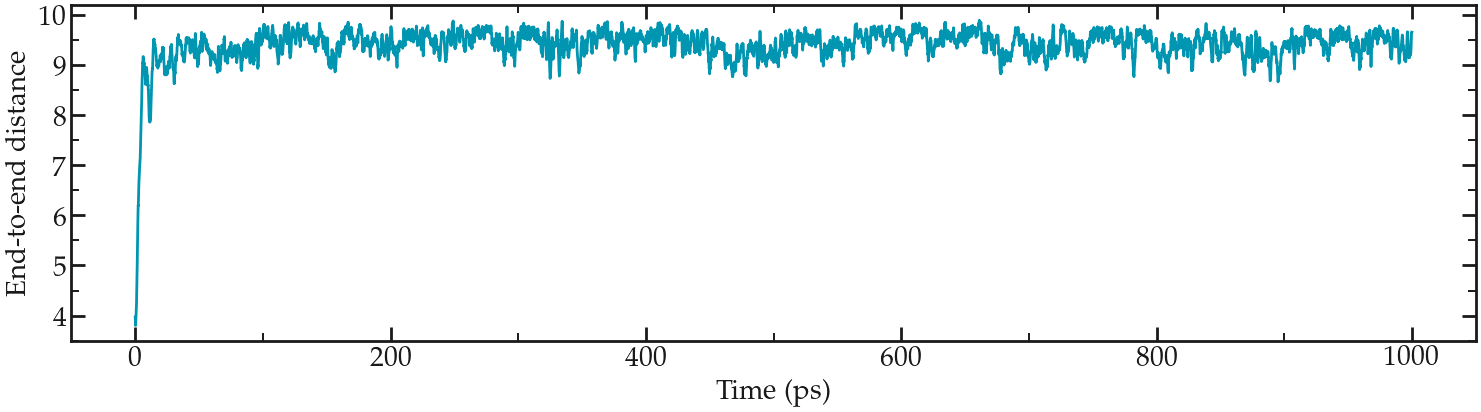
Evolution of the end-to-end distance with time.#
You can also visualize the PEG molecule during the stretching, this is what I see:

The PEG molecule under stretching in vacuum.#
PEG molecule in water#
The PEG molecule is now going to be solvated in water. Duplicate the free-peg-in-vacuum/ folder, and call the copy free-peg-in-water/.
Add water molecules to the system by using gmx solvate:
gmx solvate -cp peg.gro -cs spc216.gro -o peg_h2o.gro -p topol.top
Here spc216.gro is a default GROMACS file containing a pre-equilibrated water reservoir.
Note the differences between peg.gro and peg_h2o.gro, as well as the new line in topol.top:
[ molecules ]
; Compound #mols
PEG 1
SOL 853
Add the line #include “ff/tip3p.itp” to the topol.top file:
; Include forcefield parameters
#include "ff/charmm35r.itp"
#include "ff/peg.itp"
#include "ff/tip3p.itp
The tip3p.itp contains the information about the water molecule, and can be downloaded by clicking here, and then placed within the ff/ folder.
Support me for 1 euro per month
Become a Patreon and support the creation of content for GROMACS
Equilibrating the system#
Here we perform a 3 steps equilibration of the solvated PEG system, starting with an energy minimization, followed by a NVT run, and finally with a NPT run.
First, let us perform an energy minimization of the system:
gmx grompp -f inputs/em.mdp -c peg_h2o.gro -p topol.top -o em
gmx mdrun -deffnm em -v
The em.mdp file contains the following commands:
integrator = steep
emtol = 10
emstep = 0.0001
nsteps = 5000
nstenergy = 1000
nstxout = 100
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1
rvdw = 1
pbc = xyz
define = -DFLEXIBLE
The define = -DFLEXIBLE commands triggers the if condition within the tip3p.itp file. Therefore the water molecules behave as flexible during the minimization (rigid molecules and energy minimization usually don’t go along well). For the next steps, rigid water molecules will be used by not including this command.
Then, let us perform a NVT (constant number of particles, constant volume, constant temperature) run:
gmx grompp -f inputs/nvt.mdp -c em.gro -p topol.top -o nvt
gmx mdrun -deffnm nvt -v
Here the nvt.mdp file can be downloaded by clicking here.
Finally, perform a NPT (constant number of particles, constant pressure, constant temperature) equilibration run, using this npt file:
gmx grompp -f inputs/npt.mdp -c nvt.gro -p topol.top -o npt
gmx mdrun -deffnm npt -v
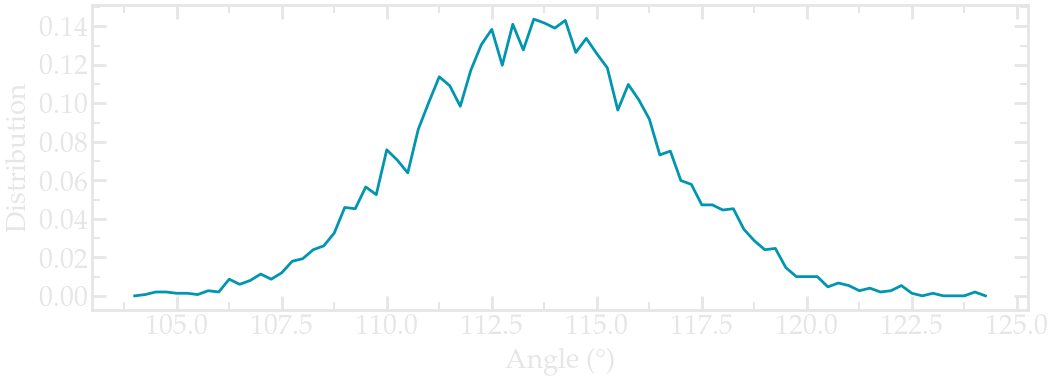
Extract the angular distribution again, and compare it to the previous vacuum simulation. Here I increased the duration of both simulations to 1 ns (for the PEG in water) and 2 ns (for PEG in vacuum) to improve the statistic (feel free to do the same if your computer allows it):
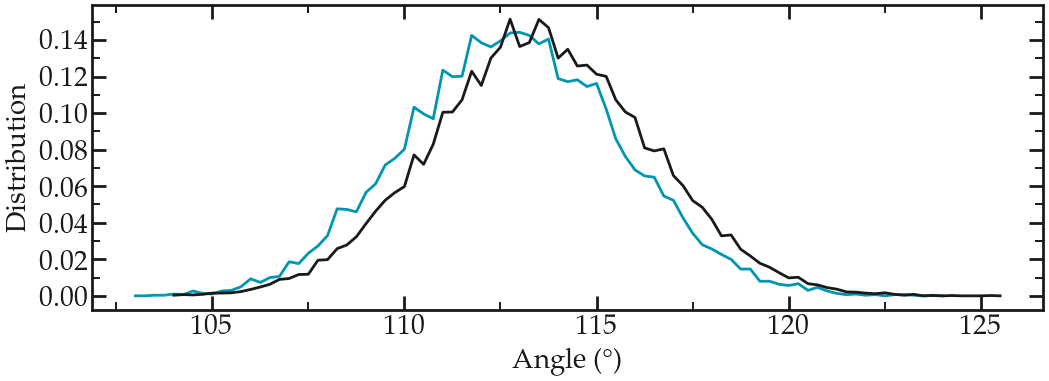
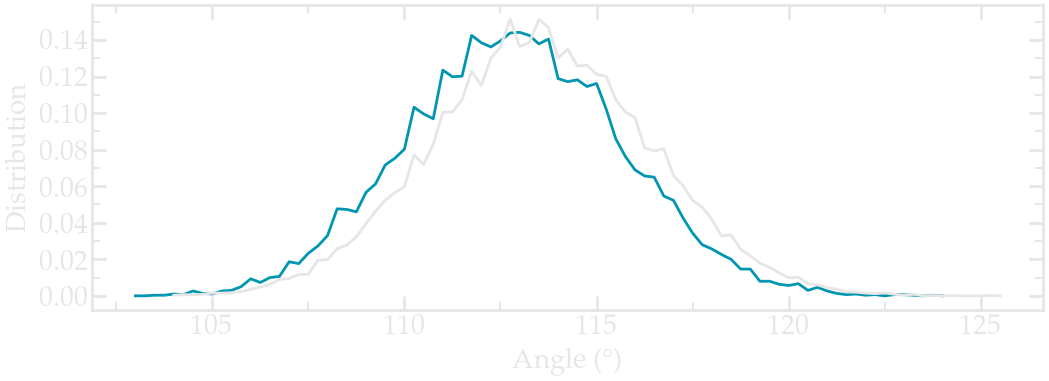
Angle distribution comparing the PEG molecule in vacuum (gray) and the PEG molecule in water (blue).#
Notice that the angle distribution is slightly shifted in water, compared to when the peg molecule is in vacuum. This indicates that the polymer has a slightly different conformation when in contact with a solvent.
Pull on the PEG#
Using the same procedure as previously, apply a force to the polymer ends and pull it inside water. Do not forget to extend the box to make space for the pulling.
Contact me
Contact me if you have any question or suggestion about these tutorials.